VYNE Therapeutics and Yarrow Bioscience Announce Merger Agreement
VYNE Therapeutics and Yarrow Bioscience Announce Merger Agreement
- Following closing, combined company plans to progress Yarrow’s lead program, YB-101, a potentially first-in-class anti-thyroid-stimulating hormone receptor (“TSHR”) antibody for Graves’ disease (“GD”) and thyroid eye disease (“TED”)
- Yarrow Bioscience is the seventh biotechnology company founded by RTW Investments and plans to advance in-licensed asset YB-101 (also known as GS-098) into a U.S.-based Phase 1b/2b trial in patients with GD in the first half of 2026; a Phase 1 trial in TED is being conducted in China by partner Changchun GeneScience Pharmaceutical Co., Ltd (“GenSci”)
- Pre-closing private financings totaling approximately $200 million expected to fund operations into 2028
- Companies to hold conference call on December 17, 2025 at 8:30AM EDT
NEW YORK, Dec. 17, 2025 (GLOBE NEWSWIRE) — VYNE Therapeutics Inc. (Nasdaq: VYNE) (“VYNE”) and Yarrow Bioscience, Inc. (“Yarrow”) today announced that they have entered into a definitive merger agreement pursuant to which the companies will combine in an all-stock transaction (the “Merger”). Upon completion of the Merger, the combined company expects to operate as Yarrow Bioscience, Inc. and trade on Nasdaq under the ticker symbol “YARW”. Following completion of the Merger, the combined company plans to focus on advancing YB-101 (also known as GS-098), a clinical-stage, potentially first-in-class TSHR antibody for the treatment of GD and TED.
In support of the Merger, a syndicate of industry-leading healthcare investors led by RTW Investments, with participation from OrbiMed, Janus Henderson Investors, venBio Partners, Logos Capital, LifeSci Venture Partners and Perceptive Advisors, has committed to pre-closing financings in Yarrow totaling approximately $200.0 million in cash proceeds.
The combined company’s cash balance at closing is expected to fund operations into 2028, including the advancement of the combined company’s lead program YB-101, into a Phase 1b/2b trial in patients with GD, which is expected to be conducted in the United States and other territories. Phase 1b data is expected in the second half of 2027. In parallel, a Phase 1 trial, which is being conducted by licensing partner GenSci, is evaluating the safety and efficacy of YB-101 in patients with TED in China.
Prior to closing, VYNE expects to declare a cash dividend to pre-Merger VYNE stockholders to distribute excess net cash, which is expected to be approximately $14.5 to $16.5 million.
“We are excited about this merger, which establishes Yarrow on a strong foundation to advance YB-101 for patients living with Graves’ disease and thyroid eye disease,” said Rebecca Frey, Pharm.D., President and Chief Executive Officer of Yarrow. “Autoimmune thyroid disorders represent areas of significant unmet need, and we believe YB-101 has the potential to deliver meaningful clinical benefit through its highly targeted TSHR-directed mechanism of action. Together with an exceptional leadership team and the support of a premier group of life sciences investors, Yarrow is well positioned to deliver on our mission to transform treatment options in this field.”
“Over the last several months, VYNE has evaluated a wide range of options to maximize stockholder value, including an assessment of our internal pipeline, financing opportunities and strategic alternatives,” said David Domzalski, President and Chief Executive Officer of VYNE. “We believe this merger provides our stockholders a compelling opportunity to realize both short- and long-term value creation through a cash dividend and the continued advancement of Yarrow’s potential breakthrough therapies for the treatment of thyroid autoimmune diseases.”
About YB-101
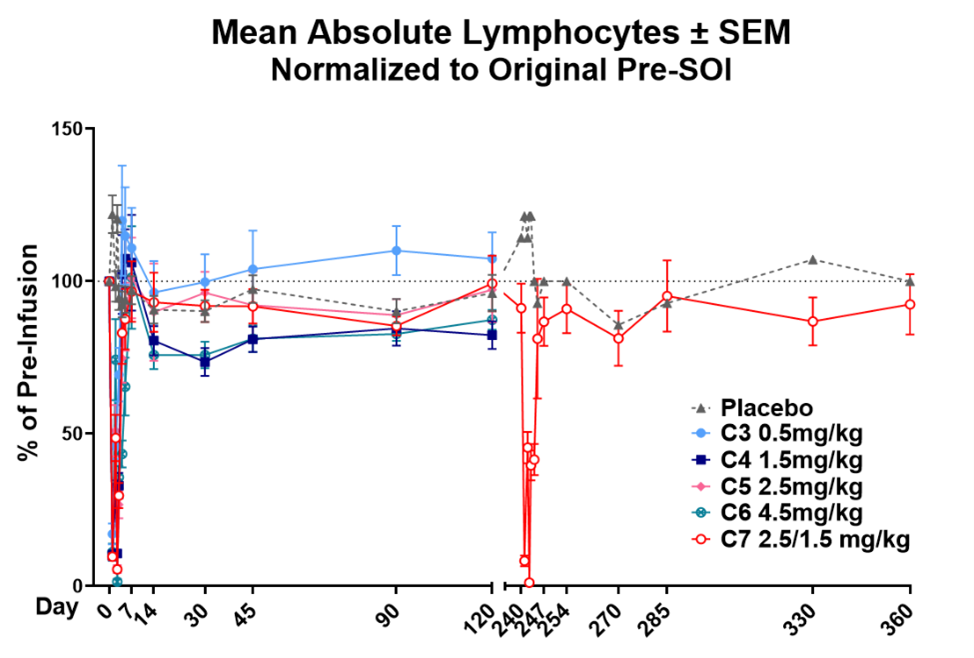
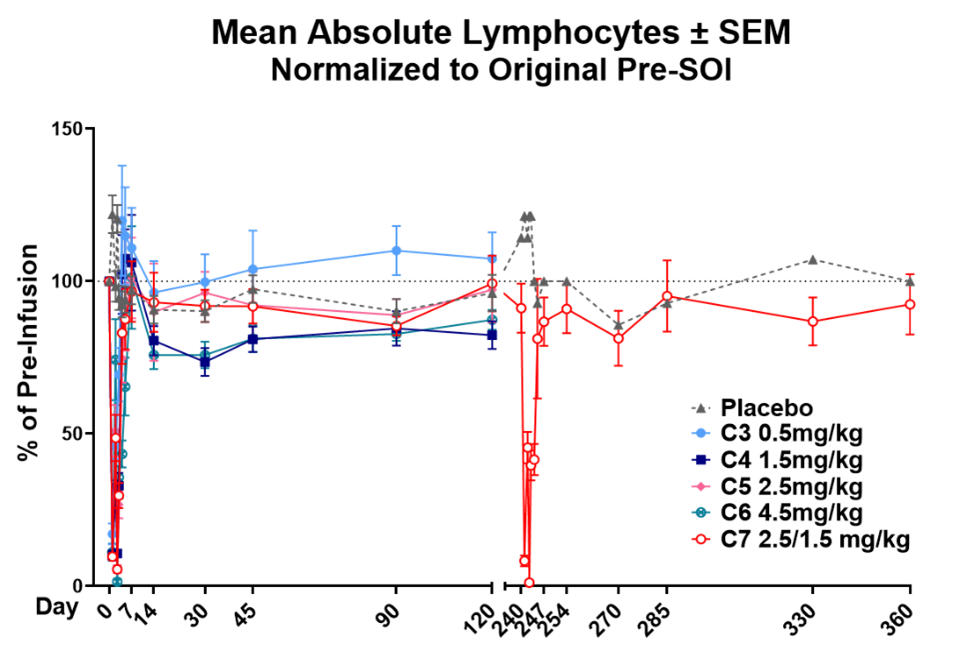
YB-101 (also known as GS-098) is a clinical-stage, humanized monoclonal antibody targeting the thyroid-stimulating hormone receptor (TSHR). The antibody is designed to rapidly and efficiently block the pathogenic activity of thyroid-stimulating autoantibodies that drive disease progression in Graves’ disease (GD) and thyroid eye disease (TED). By binding selectively to the TSH receptor and blocking autoantibody-induced receptor activation, YB-101 directly inhibits the biological pathway responsible for hyperthyroidism and orbitopathy. This novel and targeted approach represents a potential breakthrough for patients who are inadequately controlled with first-line therapies and remain at high risk for complications of GD and TED. While existing IGF-1R–directed therapies have demonstrated clinical benefit in TED, their broad receptor expression has been associated with treatment-limiting adverse events in certain patients. YB-101 targets the thyroid-stimulating hormone receptor, a key disease-driving pathway with more restricted tissue expression, and is designed to directly inhibit pathogenic autoantibody activity. This targeted approach has the potential to address an important unmet need for therapies with differentiated risk-benefit profiles. Earlier this week, Yarrow executed an exclusive license agreement with Shanghai Scizeng Medical Technology Co., Ltd, an affiliate of GenSci, to obtain global ex-China rights to develop GS-098 in GD and TED.
About the Proposed Transactions
Under the terms of the merger agreement, the pre-Merger VYNE stockholders are expected to own approximately 3% of the combined company, and the pre-Merger Yarrow stockholders (inclusive of those investors participating in the financings described above) are expected to own approximately 97% of the combined company, which is subject to adjustment in accordance with the definitive merger agreement.
The Merger has received unanimous approval by the boards of directors of both companies and is expected to close in the second quarter of 2026, subject to certain closing conditions, including, among other things, approval by the stockholders of each company, the effectiveness of a registration statement on Form S-4 to be filed with the U.S. Securities and Exchange Commission (the “SEC”) to register the securities to be issued in connection with the Merger and the satisfaction of other customary closing conditions.
The combined company will be named “Yarrow Bioscience, Inc.” and be led by Rebecca Frey, Yarrow’s Chief Executive Officer and a member of Yarrow’s board of directors. In addition, Lori Payton, Ph.D. is joining Yarrow’s management team as Chief Development Officer.
Advisory and Legal Counsel
Gibson, Dunn & Crutcher LLP is serving as legal counsel to Yarrow, while Wedbush Securities Inc. is serving as exclusive strategic financial advisor. Cooley LLP is serving as legal counsel to VYNE and LifeSci Capital is serving as exclusive financial advisor.
Conference Call Details
Yarrow and VYNE plan to hold a joint conference call on December 17, 2025 at 8:30 AM EDT to discuss the Merger in more detail. To join the webcast, please register here. A replay of the webcast can be accessed following the call by visiting www.vynetherapeutics.com.
About Yarrow Bioscience, Inc.
Yarrow Bioscience is a clinical-stage biotechnology company focused on developing transformative therapies for autoimmune thyroid diseases. The company’s lead candidate, YB-101, is a humanized monoclonal antibody targeting the thyroid-stimulating hormone receptor (TSHR) for the treatment of Graves’ disease and thyroid eye disease. Yarrow is headquartered in New York, NY, and is backed by leading healthcare investors, including RTW Investments.
For more information, please visit www.yarrowbioscience.com.
About VYNE Therapeutics Inc.
VYNE is a clinical-stage biopharmaceutical company focused on developing differentiated therapies to treat inflammatory and immune-mediated conditions with high unmet need. VYNE’s unique and proprietary BET inhibitors, which comprise its InhiBET™ platform, are designed to overcome limitations of early generation BET inhibitors by leveraging alternative routes of administration and enhanced selectivity.
For more information, please visit www.vynetherapeutics.com.
Cautionary Statement Regarding Forward-Looking Statements
Certain statements in this press release, other than purely historical information, may constitute “forward-looking statements” within the meaning of the federal securities laws, including for purposes of the safe harbor provisions under the Private Securities Litigation Reform Act of 1995. These forward-looking statements include, but are not limited to, express or implied statements regarding the structure, timing and completion of the proposed Merger; the combined companies’ cash position after closing of the proposed Merger and cash runway of the combined company; the combined company’s listing on Nasdaq after closing of the proposed Merger; expectations regarding the ownership structure of the combined company; the anticipated timing of closing; the expected executive officers of the combined company; the future operations of the combined company; the nature, strategy and focus of the combined company; the development and commercial potential and potential benefits of any product candidates of the combined company; anticipated clinical drug development activities and related timelines, including the expected timing for trial initiation, data and other clinical results; and other statements that are not historical fact. Any forward-looking statements in this release are based on management’s current knowledge and its present beliefs and expectations regarding possible future events and are subject to risks, uncertainties and assumptions that could cause actual results to differ materially and adversely from those set forth or implied by such forward-looking statements. There can be no assurance that future developments affecting the combined company will be those that have been anticipated. These forward-looking statements involve a number of risks, uncertainties (some of which are beyond the combined company’s control) or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to: the risk that the conditions to the closing of the Merger are not satisfied, including the failure to timely obtain shareholder approval for the transaction, if at all; uncertainties as to the timing of the consummation of the Merger and the ability of each of VYNE and Yarrow to consummate the Merger; risks related to VYNE’s ability to manage its operating expenses and its expenses associated with the Merger pending closing; risks related to the failure or delay in obtaining required approvals from any governmental or quasi-governmental entity necessary to consummate the Merger; the risk that as a result of adjustments to the exchange ratio, VYNE shareholders and Yarrow stockholders could own more or less of the combined company than is currently anticipated; risks related to the market price of VYNE’s common stock relative to the value suggested by the exchange ratio; unexpected costs, charges or expenses resulting from the transaction; potential adverse reactions or changes to business relationships resulting from the announcement or completion of the Merger; the uncertainties associated with Yarrow’s product candidates, as well as risks associated with the clinical development and regulatory approval of product candidates, including potential delays in the commencement, enrollment and completion of clinical trials; risks related to the inability of the combined company to obtain sufficient additional capital to continue to advance these product candidates and its preclinical programs; uncertainties in obtaining successful clinical results for product candidates and unexpected costs that may result therefrom; risks related to the failure to realize any value from product candidates and preclinical programs being developed and anticipated to be developed in light of inherent risks and difficulties involved in successfully bringing product candidates to market; risks associated with the possible failure to realize certain anticipated benefits of the Merger, including with respect to future financial and operating results; the risk that the related financing is not consummated; and the risk that VYNE’s shareholders receive more or less of the cash dividend than is currently anticipated; and those uncertainties and factors described under the heading “Risk Factors” in VYNE’s Annual Report on Form 10-K for the year ended December 31, 2024 and Quarterly Report on Form 10-Q for the quarter ended September 30, 2025, and VYNE’s other filings from time to time with the SEC. Nothing in this press release should be regarded as a representation by any person that the forward-looking statements set forth therein will be achieved or that any of the contemplated results of such forward-looking statements will be achieved. You should not place undue reliance on forward-looking statements in this press release, which speak only as of the date they are made and are qualified in their entirety by reference to the cautionary statements herein. The combined company does not undertake or accept any duty to make any updates or revisions to any forward-looking statements.
NO OFFER OR SOLICITATION
This communication is not intended to and do not constitute (i) a solicitation of a proxy, consent or approval with respect to any securities or in respect of the proposed transaction or (ii) an offer to sell or the solicitation of an offer to subscribe for or buy or an invitation to purchase or subscribe for any securities pursuant to the proposed transaction or otherwise, nor shall there be any sale, issuance or transfer of securities in any jurisdiction in contravention of applicable law. No offer of securities shall be made except by means of a prospectus meeting the requirements of the Securities Act of 1933, as amended, or an exemption therefrom. Subject to certain exceptions to be approved by the relevant regulators or certain facts to be ascertained, the public offer will not be made directly or indirectly, in or into any jurisdiction where to do so would constitute a violation of the laws of such jurisdiction, or by use of the mails or by any means or instrumentality (including without limitation, facsimile transmission, telephone and the internet) of interstate or foreign commerce, or any facility of a national securities exchange, of any such jurisdiction.
NEITHER THE SEC NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THE SECURITIES OR DETERMINED IF THIS COMMUNICATION IS TRUTHFUL OR COMPLETE
IMPORTANT ADDITIONAL INFORMATION ABOUT THE PROPOSED TRANSACTION WILL BE FILED WITH THE SEC
This communication is not a substitute for any other document that VYNE may file with the SEC in connection with the proposed transaction, including the registration statement on Form S-4 (the “Form S-4”) that will contain a proxy statement and prospectus. In connection with the proposed transaction between VYNE, Yarrow and Merger Sub, VYNE intends to file relevant materials with the SEC, including the Form S-4. VYNE URGES INVESTORS AND STOCKHOLDERS TO READ THE REGISTRATION STATEMENT, INCLUDING THE PROXY STATEMENT/PROSPECTUS CONTAINED THEREIN, AND ANY OTHER RELEVANT DOCUMENTS THAT MAY BE FILED WITH THE SEC, AS WELL AS ANY AMENDMENTS OR SUPPLEMENTS TO THESE DOCUMENTS, CAREFULLY AND IN THEIR ENTIRETY IF AND WHEN THEY BECOME AVAILABLE BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION ABOUT VYNE, YARROW, MERGER SUB, THE PROPOSED TRANSACTION AND RELATED MATTERS. Investors and stockholders will be able to obtain free copies of the Form S-4 and other documents filed by VYNE with the SEC (when they become available) through the website maintained by the SEC at www.sec.gov. In addition, investors and stockholders should note that VYNE communicates with investors and the public using its website (www.vynetherapeutics.com) and the investor media website (https://vynetherapeutics.com/investors-media) where anyone will be able to obtain free copies of the Form S-4 and included proxy statement/prospectus and other documents filed by VYNE with the SEC and stockholders are urged to read the Form S-4 and included proxy statement/prospectus and the other relevant materials when they become available before making any voting or investment decision with respect to the proposed transaction.
PARTICIPANTS IN THE SOLICITATION
VYNE, Yarrow and their respective directors and executive officers may be deemed to be participants in the solicitation of proxies from stockholders in connection with the proposed transaction. Information about VYNE’s directors and executive officers, including a description of their interests in VYNE, is included in VYNE’s most recent definitive proxy statement, as filed with the SEC on November 12, 2025. Additional information regarding these persons and their interests in the proposed transaction will be included in the proxy statement/prospectus relating to the proposed transaction when it is filed with the SEC. These documents can be obtained free of charge from the sources indicated above.
Third-party products and company names mentioned herein may be the trademarks of their respective owners.
Yarrow Media Contact:
Ten Bridge Communications
TBCYarrow@tenbridgecommunications.com
VYNE Investor Relations:
John Fraunces
LifeSci Advisors, LLC
jfraunces@lifesciadvisors.com