
Merck’s Clesrovimab (MK-1654), an Investigational Respiratory Syncytial Virus (RSV) Preventative Monoclonal Antibody, Significantly Reduced Incidence of RSV Disease and Hospitalization in Healthy Preterm and Full-term Infants
Merck’s Clesrovimab (MK-1654), an Investigational Respiratory Syncytial Virus (RSV) Preventative Monoclonal Antibody, Significantly Reduced Incidence of RSV Disease and Hospitalization in Healthy Preterm and Full-term Infants

In the Phase 2b/3 trial, clesrovimab reduced RSV-associated hospitalizations (secondary endpoint) and RSV-associated lower respiratory infection hospitalizations (tertiary endpoint) by more than 84% and 90%, respectively, through 5 months
Clesrovimab has the potential to become the first and only approved immunization designed to protect infants with the same single dose regardless of weight for the duration of their first RSV season
RAHWAY, N.J.–(BUSINESS WIRE)–$MRK #MRK–Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced the presentation of positive results from the Phase 2b/3 clinical trial (MK-1654-004) evaluating clesrovimab, the company’s investigational prophylactic monoclonal antibody designed to protect infants from respiratory syncytial virus (RSV) disease during their first RSV season. The results, along with interim findings from the ongoing Phase 3 trial (MK-1654-007) of clesrovimab, were presented during IDWeek 2024, held October 16-19 in Los Angeles, California.
![]()
![]()
Results from MK-1654-004, a placebo-controlled Phase 2b/3 pivotal trial evaluating a single dose of clesrovimab administered to healthy preterm and full-term infants (birth to 1 year of age) met all prespecified endpoints, with consistent results through both the 5-month and 6-month time points. The incidence of adverse events (AEs) and serious AEs were comparable between the clesrovimab and placebo groups, and there were no treatment or RSV-related deaths during the study.
“RSV continues to be a widespread seasonal infection that can affect both healthy and at-risk infants and is the leading cause of hospitalization for infants,” said Dr. Octavio Ramilo, chair of the Department of Infectious Diseases at St. Jude’s Children’s Research Hospital and investigator for the MK-1654-004 and MK-1654-007 trials. “The MK-1654-004 study evaluated a broad spectrum of RSV disease ranging from mild outpatient illness to severe disease requiring hospitalization. These promising results demonstrating decreased incidence of RSV disease, including hospitalizations, highlight the potential for clesrovimab to play an important role in helping to alleviate the continued burden of RSV on infants and their families.”
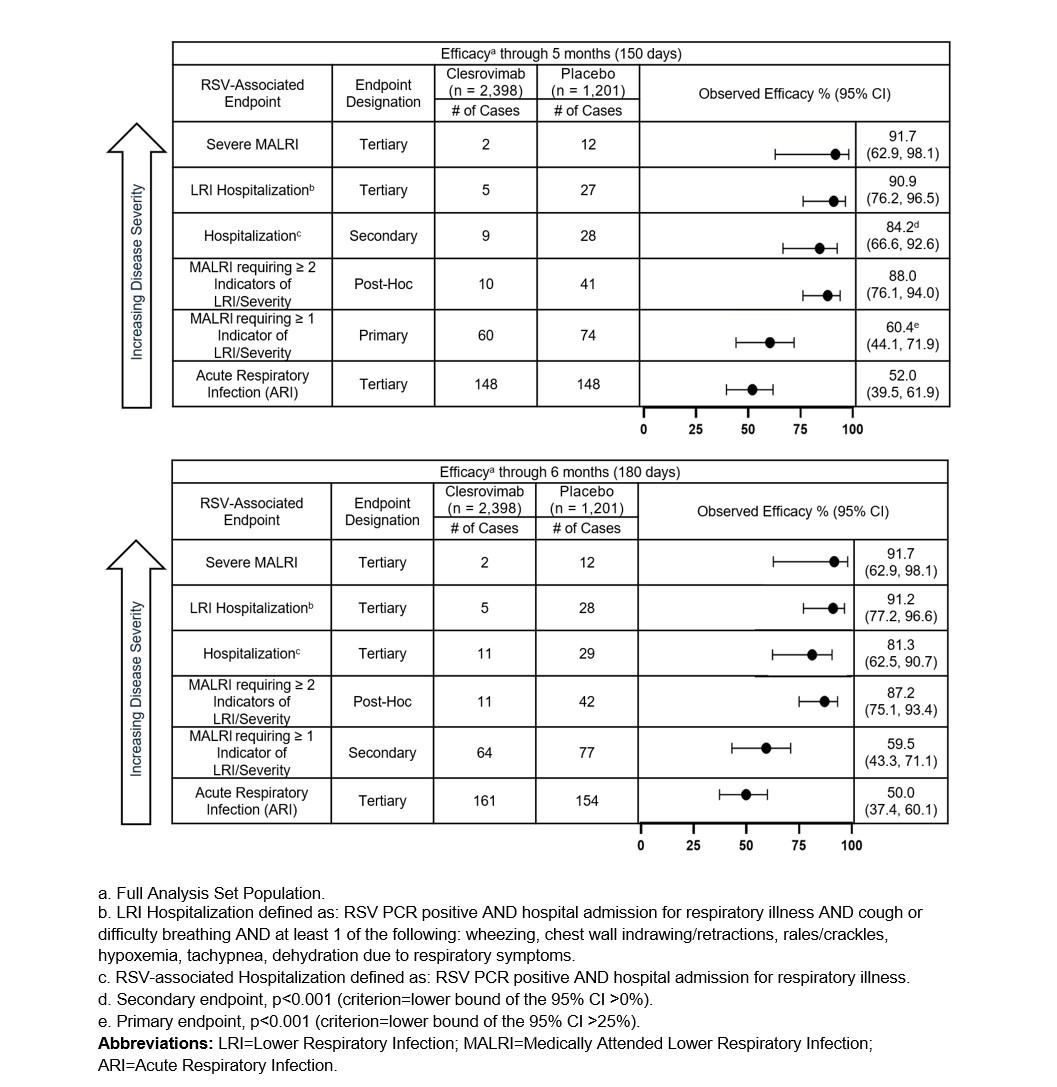
The primary efficacy endpoint of the trial, the reduction in incidence of RSV-associated medically attended lower respiratory infections (MALRI) requiring ≥ 1 indicator of lower respiratory infection (LRI) or severity compared to placebo through Day 150 (5 months) postdose, was 60.4% (95% CI: 44.1, 71.9, p<0.001). Clesrovimab also reduced RSV-associated hospitalizations (secondary endpoint) and RSV-associated LRI hospitalizations (tertiary endpoint) through Day 150 (5 months) compared to placebo by 84.2% (95% CI: 66.6, 92.6, p<0.001) and 90.9% (95% CI: 76.2, 96.5), respectively. Clesrovimab reduced the incidence of severe MALRI (tertiary endpoint) by 91.7% (95% CI: 62.9, 98.1).
In addition, in a post hoc analysis, the reduction in incidence of MALRI requiring ≥ 2 indicators of LRI and severity (an endpoint of more severe MALRI than the primary MALRI endpoint), was 88.0% (95% CI: 76.1, 94.0) through Day 150 (5 months).
Additional details on the data from the MK-1654-004 trial across RSV disease burden are presented in order of decreasing disease severity endpoints in Figure 1 above.
Merck also announced data from a planned interim analysis of the MK-1654-007 trial, a Phase 3 trial evaluating the safety and efficacy of clesrovimab versus palivizumab in infants and children at increased risk for severe RSV disease. The primary endpoint of the study is the safety and tolerability of clesrovimab in infants entering their first RSV season. Interim results showed clesrovimab had a comparable safety profile to palivizumab, and no drug-related serious AEs were reported to date. Incidence rates of RSV-associated MALRI requiring ≥ 1 indicator of LRI or severity and RSV-associated hospitalizations (secondary endpoints) were also comparable between clesrovimab (3.6% and 1.3%, respectively) and palivizumab (3.0% and 1.5%, respectively) through Day 150 (5 months).
“The breadth of data presented at IDWeek highlight the potential for clesrovimab to help lessen the significant impact RSV can have on infants and their families, as well as the strain on healthcare systems due to high infection and hospitalization rates,” said Dr. Paula Annunziato, senior vice president, infectious diseases and vaccines, Global Clinical Development, Merck Research Laboratories. “These clinically meaningful findings also reinforce the potential for clesrovimab to be the first and only immunization designed to protect both healthy and at-risk infants using the same dose, regardless of weight. We look forward to continuing to discuss these data with health authorities around the world with the goal of making clesrovimab available for infants as early as the 2025-26 RSV season.”
About MK-1654-004
MK-1654-004 (NCT04767373) is a Phase 2b/3 double-blind, randomized, placebo-controlled clinical trial to evaluate the safety and efficacy of clesrovimab in healthy preterm and full-term infants from birth to 1 year of age entering their first RSV season. The study enrolled 3,632 participants who were randomized 2:1 to receive either a single fixed dose of clesrovimab (105 mg intramuscular injection (IM)) or placebo on Day 1. Primary endpoints included the incidence of participants with RSV-associated medically attended lower respiratory infection (MALRI) from Day 1 (postdose) to Day 150 as compared to placebo and safety. The MALRI definition required >1 indicator of LRI or severity. RSV-associated hospitalization through Day 150 and MALRI requiring >1 indicator of LRI or severity to Day 180, were prespecified secondary endpoints. Prespecified tertiary endpoints included acute respiratory infection, RSV-associated lower respiratory infection hospitalizations and incidence of severe MALRI through Day 150. In a post hoc analysis, more severe forms of RSV-associated MALRI (>2 indicators of LRI and severity) were assessed. Across endpoints, additional measures of efficacy were assessed through Day 180. Safety measures included the percentage of participants with solicited injection-related adverse events (AEs), AEs of special interest (AESIs) solicited systemic AEs or serious adverse events (SAEs).
About MK-1654-007
MK-1654-007 (NCT04938830) is a Phase 3, multicenter, randomized, partially blinded, controlled study to evaluate the safety, efficacy, and pharmacokinetics of clesrovimab in infants and children at increased risk for severe RSV disease compared to palivizumab. The study enrolled participants who were entering their first RSV season and recommended to receive palivizumab due to prematurity (≤35 weeks gestational age), chronic lung disease (CLD) of prematurity, or hemodynamically significant congenital heart disease (CHD). Participants were randomized 1:1 to receive clesrovimab (105 mg IM on Day 1, placebo on Day 28) or monthly palivizumab in their first season, and eligible participants received clesrovimab (210 mg IM) in the second RSV season. At this interim analysis, 901 participants were enrolled in the trial. The primary endpoint is safety and tolerability of clesrovimab versus palivizumab in the first season. Secondary endpoints include the incidence of RSV-associated medically attended lower respiratory infections (MALRI) requiring ≥1 indicator of LRI or severity and of RSV-associated hospitalization through Day 150.
About clesrovimab (MK-1654)
Clesrovimab (MK-1654) is an investigational, extended half-life monoclonal antibody (mAb) developed as a passive immunization for the prevention of RSV. Clesrovimab is designed to be administered as the same single dose, regardless of birth weight, and is being studied in healthy preterm, full-term and at-risk infants to provide direct, rapid, and durable protection through their first RSV season against mild, moderate and severe RSV.
About RSV
Respiratory syncytial virus (RSV) is a contagious virus that causes widespread seasonal infections like the flu, with a worldwide burden in infants and older adults. There is high unmet need for preventative options in both healthy and high-risk infants. Globally, RSV is the leading cause of hospitalization for healthy infants under a year old. RSV can lead to serious respiratory conditions like bronchiolitis and pneumonia, causing an estimated 101,000 deaths a year worldwide in children under five. According to the CDC, RSV season starts in the fall and peaks in the winter in most regions of the United States, but timing and severity in a given community or region can vary year to year.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on X (formerly Twitter), Facebook, Instagram, YouTube and LinkedIn.
Forward-Looking Statement of Merck & Co., Inc., Rahway, N.J., USA
This news release of Merck & Co., Inc., Rahway, N.J., USA (the “company”) includes “forward-looking statements” within the meaning of the safe harbor provisions of the U.S. Private Securities Litigation Reform Act of 1995. These statements are based upon the current beliefs and expectations of the company’s management and are subject to significant risks and uncertainties. There can be no guarantees with respect to pipeline candidates that the candidates will receive the necessary regulatory approvals or that they will prove to be commercially successful. If underlying assumptions prove inaccurate or risks or uncertainties materialize, actual results may differ materially from those set forth in the forward-looking statements.
Risks and uncertainties include but are not limited to, general industry conditions and competition; general economic factors, including interest rate and currency exchange rate fluctuations; the impact of pharmaceutical industry regulation and health care legislation in the United States and internationally; global trends toward health care cost containment; technological advances, new products and patents attained by competitors; challenges inherent in new product development, including obtaining regulatory approval; the company’s ability to accurately predict future market conditions; manufacturing difficulties or delays; financial instability of international economies and sovereign risk; dependence on the effectiveness of the company’s patents and other protections for innovative products; and the exposure to litigation, including patent litigation, and/or regulatory actions.
The company undertakes no obligation to publicly update any forward-looking statement, whether as a result of new information, future events or otherwise. Additional factors that could cause results to differ materially from those described in the forward-looking statements can be found in the company’s Annual Report on Form 10-K for the year ended December 31, 2023 and the company’s other filings with the Securities and Exchange Commission (SEC) available at the SEC’s Internet site (www.sec.gov).
Contacts
Media:
Julie Cunningham
(617) 519-6264
Kimberly Petrillo
(267) 742-2813
Investors:
Alexis Constantine
(732) 594-1578
Peter Dannenbaum
(732) 594-1579





